

青岛能源所揭示双原子簇催化水分解反应机理
原子精确的团簇催化剂作为均相和非均相催化之间的桥梁,可以用于深入解析催化反应构-效关系。然而目前纳米团簇催化剂的非单一活性位点之间的协同作用机制鲜有报道。为此,青岛能源所团簇化学与能源催化研究组针对电解水制氢过程,首次深入探讨了双活性中心在水分解过程中氧耦合机理起到的关键作用,为电解水制氢过程提供有效的理论依据。研究利用高通量密度泛函理论,以碳载金属双原子簇为模型催化剂,发现7种异核和4种同核双原子催化剂性能优于已报道的最低理论过电位,并合理设计了用于活性预测的描述符,这也为团簇催化剂活性中心以及相应反应机理的研究奠定了理论基础。成果在线发表于Nature Communications。
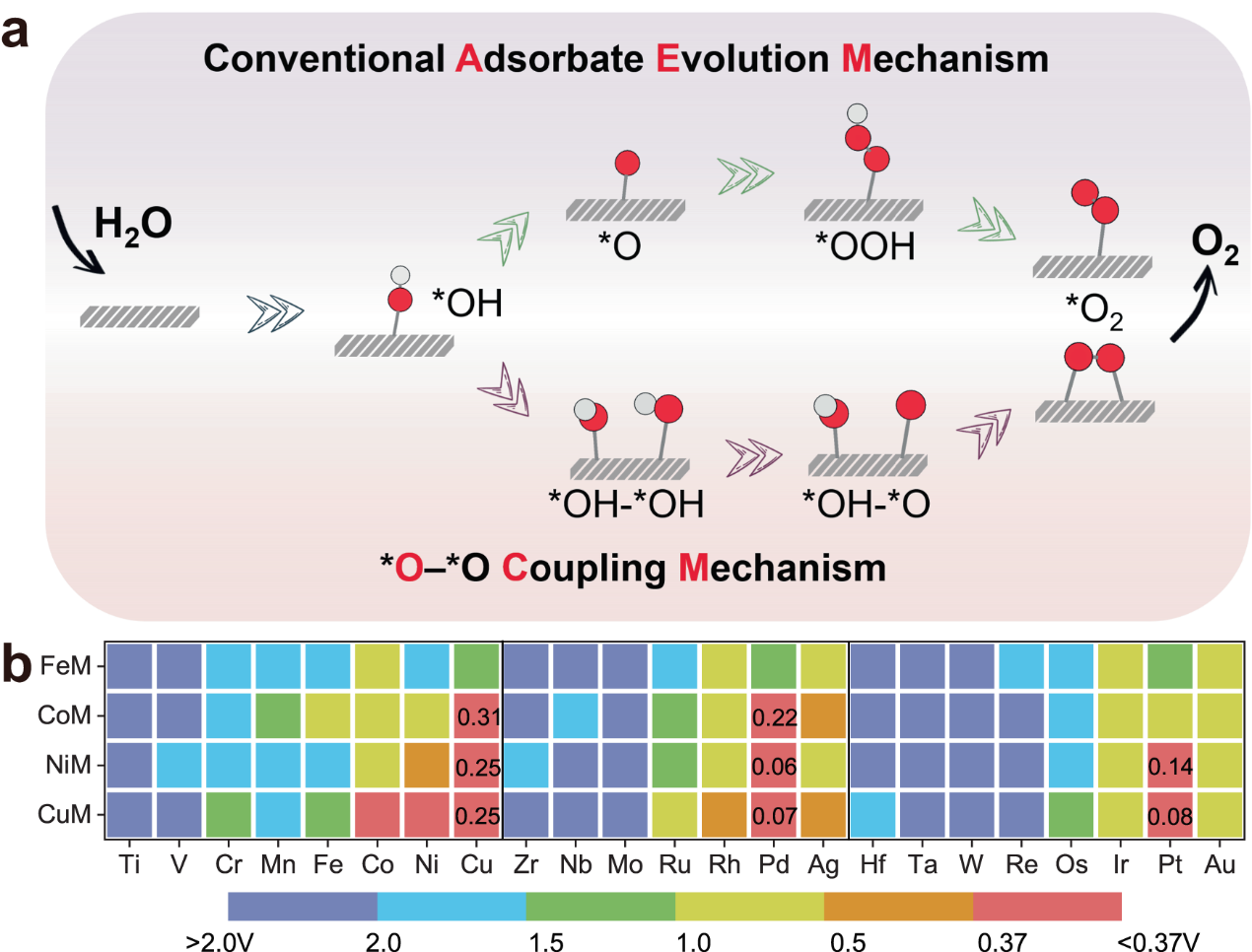
图1. (a)传统吸附演化机理(AEM)以及氧耦合机理(OCM)的反应路径示意图,(b)遵循OCM路径的82种FeM、CoM、NiM和CuM催化剂的OER活性地图。
研究人员受到氢氧化物的晶格氧机理(LOM)的启发,氧耦合机理(OCM)也同样是两个相邻活性中心上吸附的O原子耦合形成产物O2 (图1a)。传统的吸附演化机理(AEM)受限于单一活性位点上,关键含氧中间体(*OH、*O和*OOH)的结合能之间的比例关系,从而导致催化剂活性无法突破理论最低过电位0.37 V。然而通过预选择的OCM路径对82种异核碳载金属双原子簇(M'M@NC)进行水氧化(OER)活性筛选,绘制出的活性地图显示这种非传统机理可以使OER活性提高(图1b),尤其是NiPd@NC(η= 0.06 V)、CuPd@NC(η= 0.07 V)和CuPt@NC(η= 0.08 V)。进一步的分析发现,活性的提高归因于双金属活性位之间的协同能够改变关键含氧中间体吸附(*OH、*OH-*OH、*OH-*O和 *O2),规避由传统AEM机理的关键含氧中间体所带来的比例关系限制,进而突破最低理论过电位0.37 V的瓶颈(图2)。
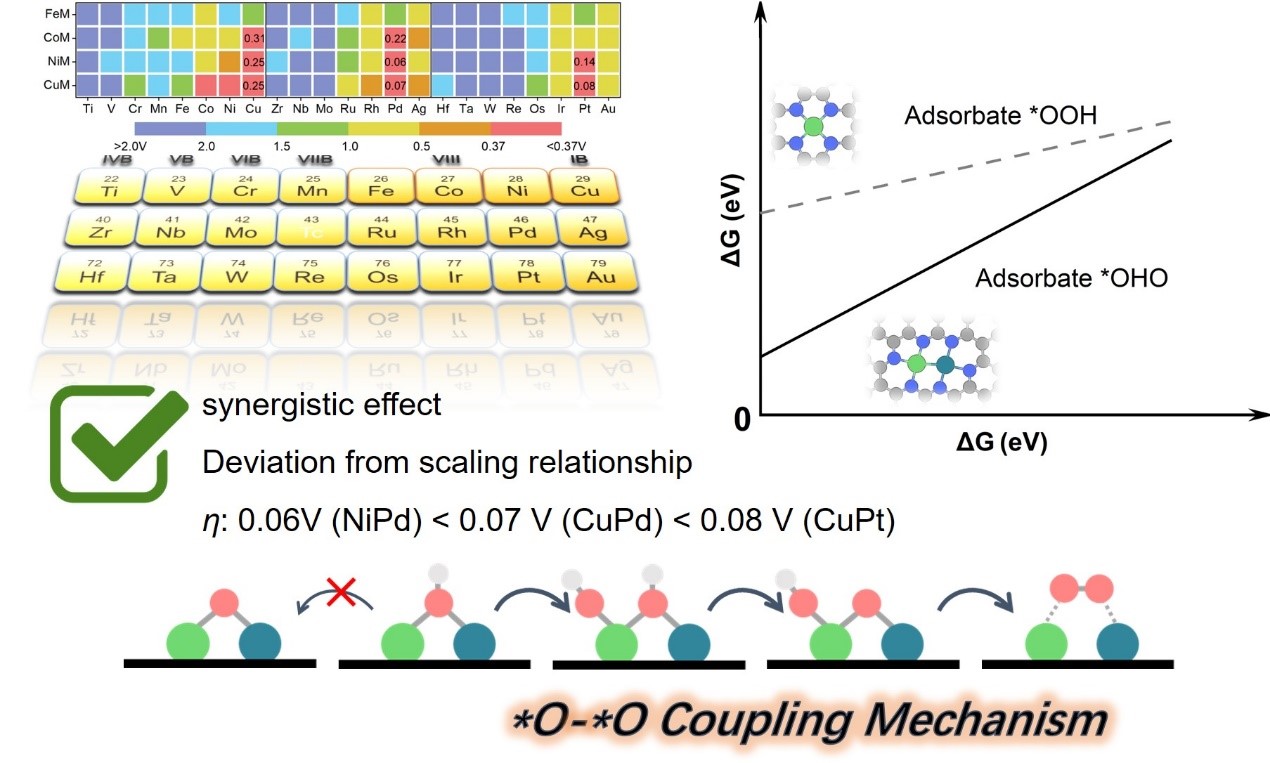
图2. 氧耦合机理使得关键含氧物种吸附能之间的比例关系发生偏移,进而显著提高反应活性。
为了使该研究更具有普适性,通过合理设计获得的描述符ΔG(*OH-*OH)可以用来预测反应活性,从而为其他相关研究节省计算资源以及缩短开发时间。经过验证,该描述符可以准确预测出高活性的同核催化剂MM@NC,同时也确保了该描述符以及设计策略的有效性。在此基础上,研究人员还根据吉布斯自由能垒作为判据,以判断两种不同机理的竞争反应(*OH→*OH–*OH vs. *OH→*O)发生的可能性,并辅以电子结构计算验证。以上内容的计算方法是基于恒电荷方法的计算氢电极模型,为了使理论结果更加贴合于真实电化学测试条件,研究人员还采用了基于巨正则系综的恒电势方法对高活性的三种催化剂(NiPd@NC、CuPd@NC和CuPt@NC)进一步考察了外加电势以及pH对反应活性的影响(图3)。研究结果表明,一方面每一个基元步骤所需要的自由能变会随着外加电势的不断调整而改变,另一方面不同的pH条件也会引起势能控制步骤(PDS)的改变,同时上述三种催化剂在酸性和碱性条件下都是非常有前途的候选催化剂。上述研究不仅为团簇催化剂的非单一活性位点提高反应性能提供基本见解,还有助于为包括OER在内的其他电化学反应催化剂的合理设计提供理论指导。
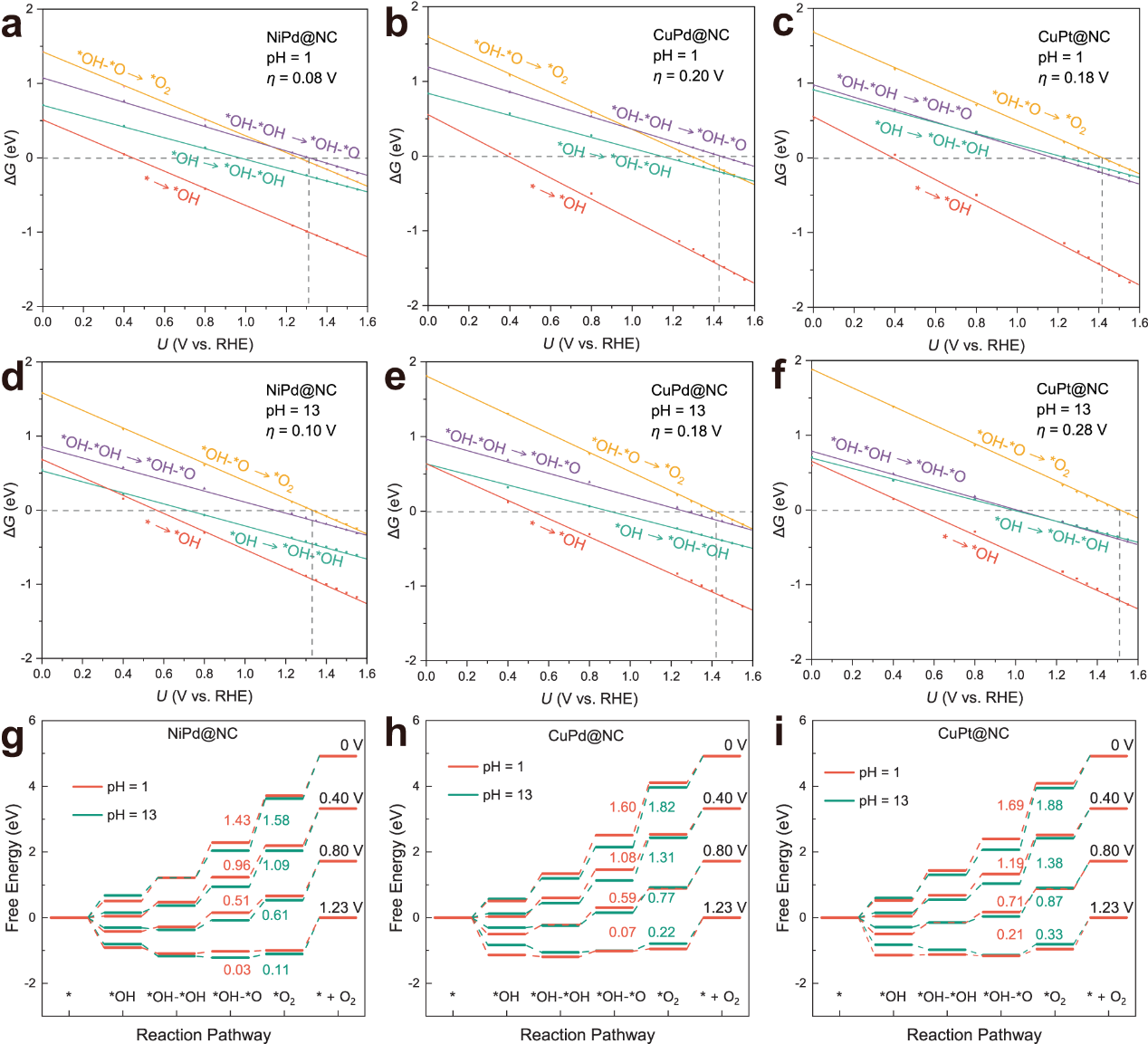
图3. 基于恒电势方法的NiPd@NC、CuPd@NC和CuPt@NC的自由能图。
该研究由团簇化学与能源催化研究组孙晓岩研究员带领完成,科研助理房聪为论文的第一作者,孙晓岩研究员和中国科学院兰州化学物理研究所丁玉晓研究员为通讯作者。该工作得到了山东能源研究院科研创新基金和山东省自然科学基金等项目的支持。(文/图 房聪)
原文链接: www.nature.com/articles/s41467-023-40177-1
Cong Fang, Jian Zhou, Lili Zhang, Wenchao Wan, Yuxiao Ding*, Xiaoyan Sun*. Synergy of Dual-atom Catalysts Deviated from the Scaling Relationship for Oxygen Evolution Reaction. Nat. Commun. 14, 4449 (2023).
附件下载:





