

青岛能源所发展气相团簇质谱技术解析单原子催化剂配位环境
催化是现代工业的重要基石,面临着精准设计和优化复杂催化体系的核心挑战,这一挑战的关键在于深入理解催化剂活性位点的结构特征及其构效关系。以氮掺杂碳材料负载单原子催化剂(M-N-C SACs)为例,尽管其在催化领域展现出巨大潜力,但关于配位环境如何调控反应活性的机制仍存在争议。这些争议主要源于两方面:一是受合成条件的影响,实际催化剂中不可避免地存在活性位点的不均一性及团簇聚集体的干扰,导致理论模型与真实体系存在差异;二是现有表征手段,如X射线吸收谱、电子显微镜等,难以在原子分辨率下精确解析单原子的配位结构及其中间体的吸附特性。因此,开发新方法以在分子水平上揭示M-N-C SACs的构效关系,是当前推动其精准设计和性能优化的迫切需求。
气相团簇模型方法在研究单原子催化剂的构效关系方面具有独特优势。与实际催化体系相比,团簇模型具有原子数精确可控、组成结构明确、电子状态清晰等特点,其构效关系能够通过气相离子-分子反应实验结合高精度量子化学计算实现分子水平的精准解析。M-N-C SACs的活性位点由孤立金属原子及其局域配位环境共同构成,这种配位模式与单金属配合物高度相似。针对不同配位结构碳载单原子构效关系不清的问题,青岛能源所团簇化学与能源催化团队联合兰州化物所丁玉晓研究员,提出了“碎片化解耦分析”的策略,即通过构建配位环境明确的气相单金属团簇模型来解耦不同M-N-C SACs活性位点对催化性能的本征贡献。这一方法能够规避实际催化体系中合成方法和表征手段的局限性,利用团队自主改造的线性离子阱质谱,在分子水平上实现了不同配位构型本征活性的精准解析。
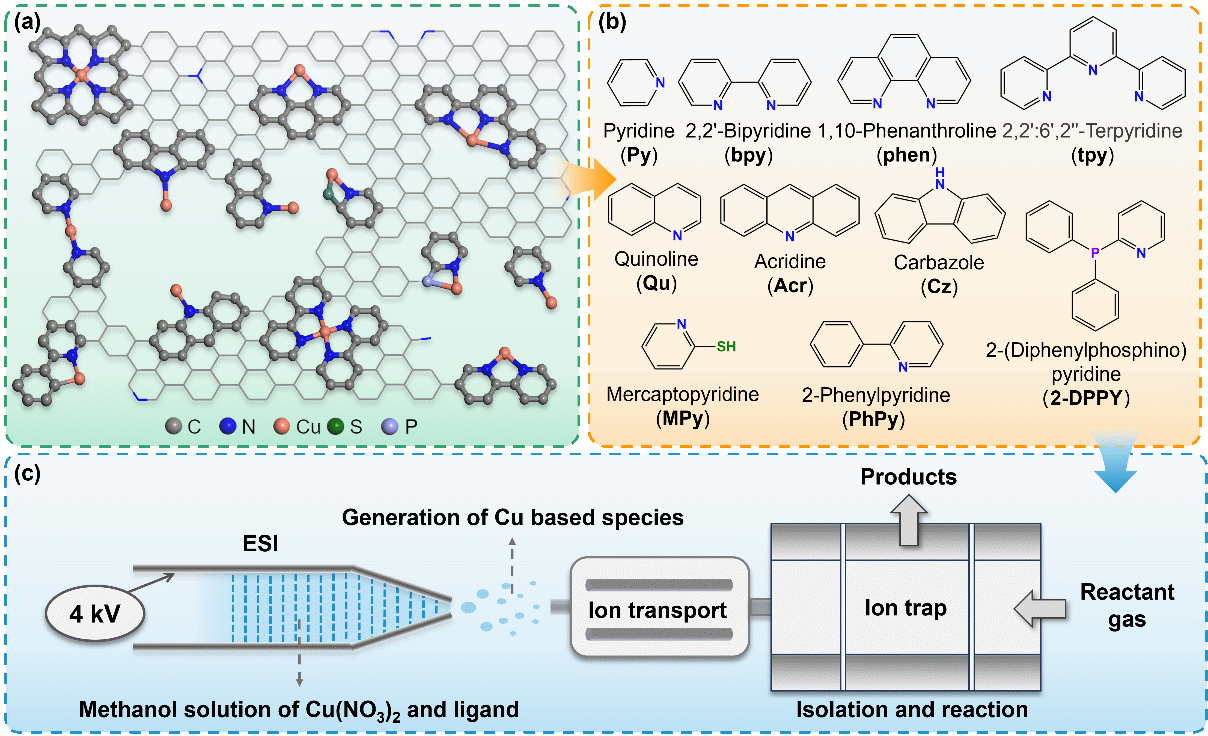
图1 Cu(I)-N-C 模型团簇生成与反应装置示意图
本研究以CO吸附作为探针反应,系统揭示了Cu-N-C SACs体系中不同N配位数(Cu-Nx,x = 0-4)、N配位构型、共掺杂原子以及配体尺寸对CO吸附活化本征活性的调控规律。其中,N配位数降低导致Cu中心电荷量升高,显著增强了CO吸附强度;同时,配体的共轭效应能够提高Cu中心与CO成键轨道间的能量匹配,进而促进CO的吸附动力学过程。此外,N配位构型也可以调控Cu中心电荷态,从而显著影响CO吸附性能。例如,对位双吡啶构型引起Cu中心电荷量的降低,导致CO吸附能力减弱;吡咯N配位则更容易稳定高价态的Cu中心。针对掺杂体系,邻位碳改性(P/S掺杂、苯基基团引入)通过重构成键轨道、调整电荷分布或扩展共轭体系等方式调控反应性能。并且,碳载体锯齿(zig-zag)边缘的共轭扩展可以提高成键轨道间的能量匹配并降低Cu中心电荷量,在提升吸附速率的同时降低成键强度;而扶手椅(armchair)边缘的影响则相对有限。这些发现为理解Cu-N-C SACs催化性能的调控机制提供了理论依据。
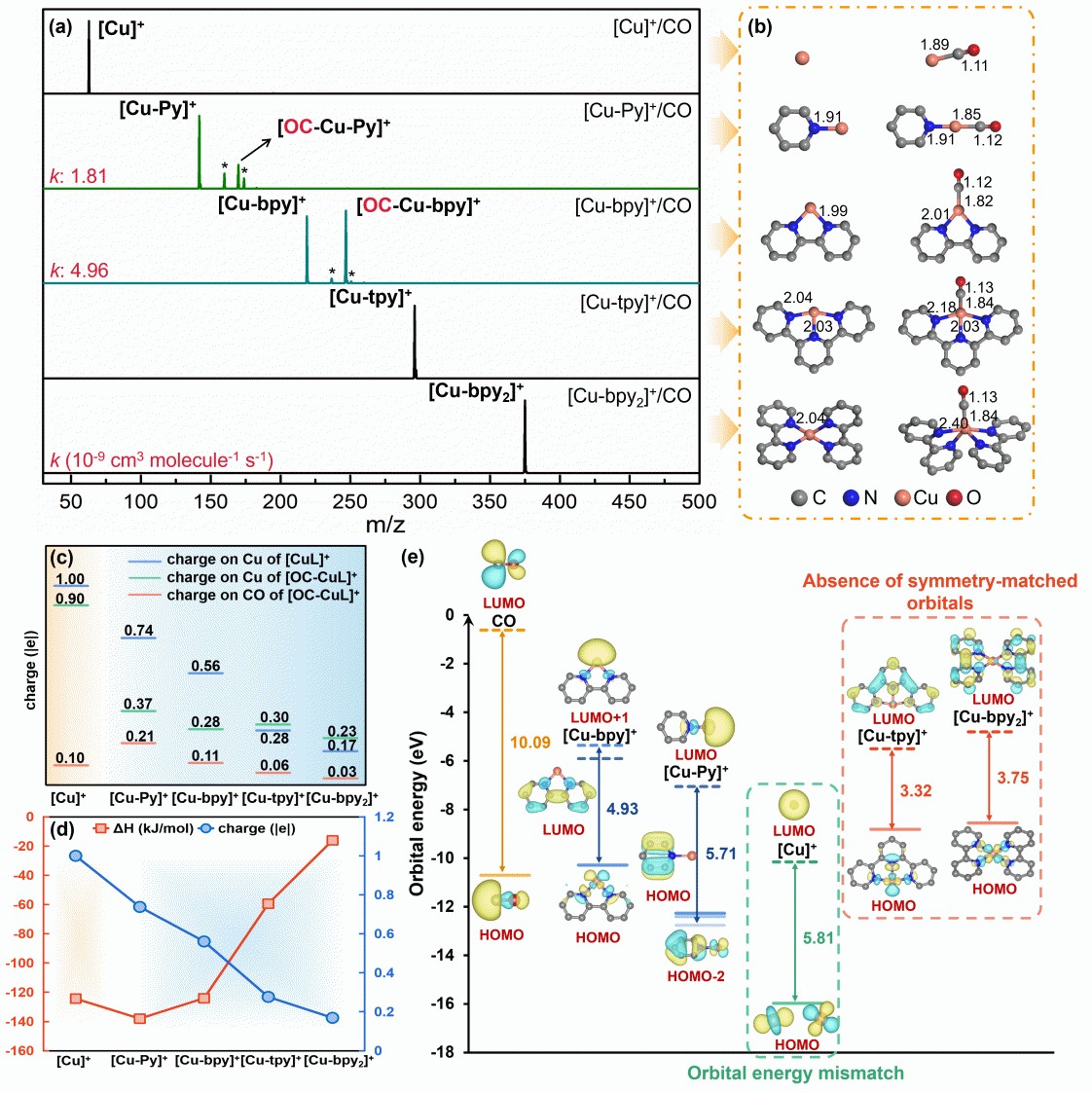
图2 不同N配位数目的团簇模型及其反应活性对比
结合上述大量具有明确配位构型的团簇模型的反应特征,研究团队进一步总结出了普适性规律:Cu中心的电荷量可以作为调控Cu-N-C活性位点CO吸附强度的热力学描述符,而Cu-N-C前线轨道能隙作为CO吸附速率的动力学描述符(图3a-b)。此外,该方法也适用于其他探针分子(如N2和C2H4吸附活化等)以及不同金属中心类型(如Co等),实验观察到了与Cu中心吸附CO行为显著不同的现象,验证了该方法的普适性。
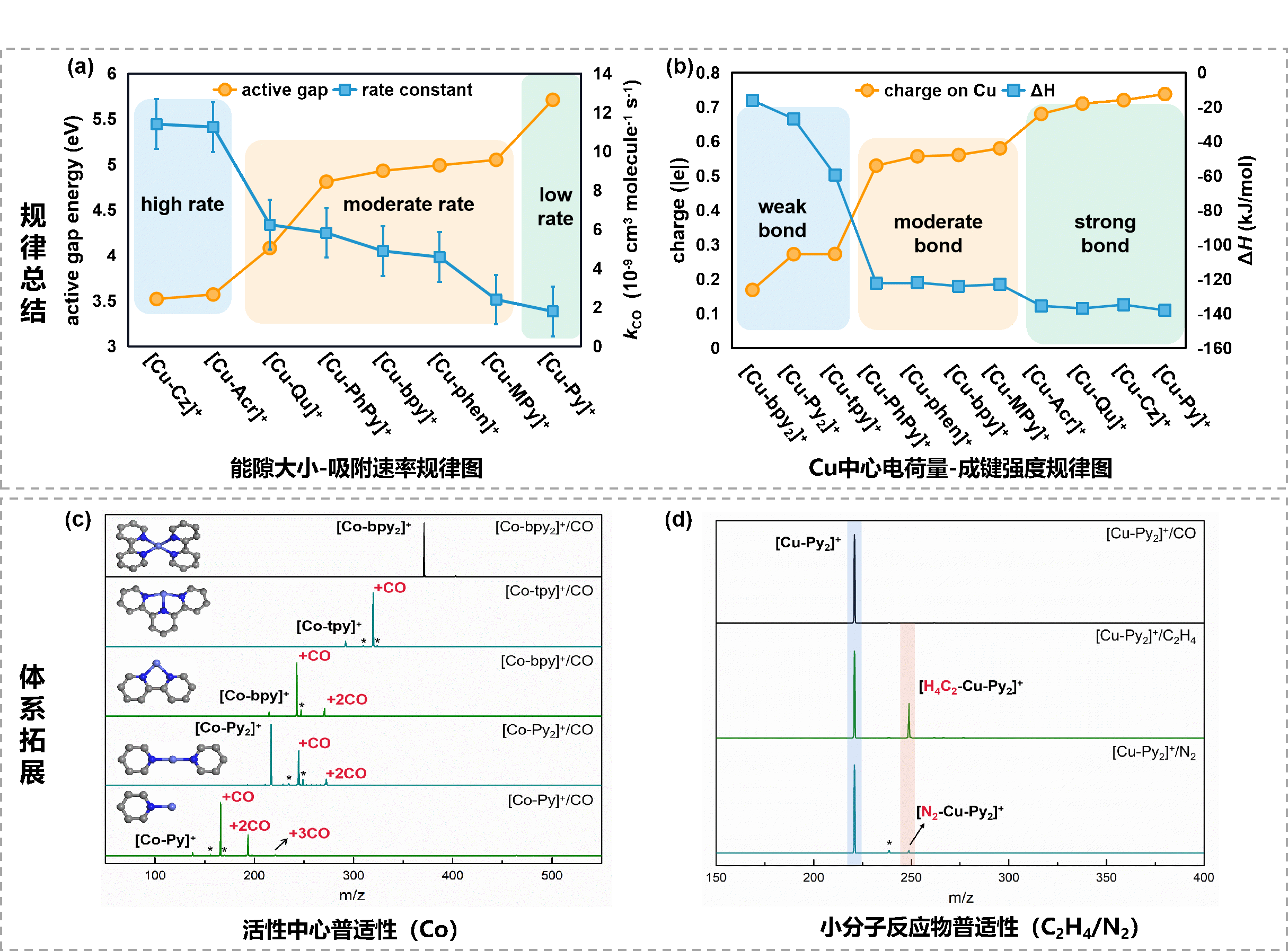
图3 反应规律总结及体系拓展情况
该工作具有以下创新点:
1.利用“碎片化解耦策略”,通过实验手段在分子水平上首次精确解析了Cu-N-C SACs体系中N配位数、N配位构型、S和P掺杂以及配体尺寸对CO吸附本征活性的影响。
附件下载:





